

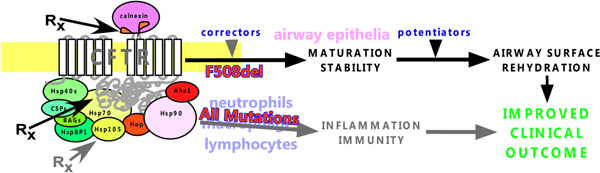
Abstract
Cystic Fibrosis (CF) is largely caused by protein misfolding and the loss of function of a plasma membrane anion channel known as the cystic fibrosis transmembrane conductance regulator (CFTR). The most common CF-causing mutation, F508del, leads to severe conformational defect in CFTR. The cellular chaperone machinery plays an important role in CFTR biogenesis and quality control. Multiple attempts have been made to improve the cell surface functional expression of the mutant CFTR by modulating the expression of components of the cellular chaperone machinery. The efficacy of such an approach has been low largely due to the severe intrinsic folding defects of the F508del CFTR. Moreover, the impact of chaperone perturbation on the chaperone machinery itself and on other physiologically important proteins might lead to potentially severe side effects. Approaches aimed at disrupting chaperone-CFTR interactions show greater efficacy, and are compatible with small-molecule drug discovery and gene therapy. Combination between chaperone modulators and F508del correctors might further enhance potency and specificity. As molecular chaperones play important roles in regulating inflammation and immunity, they can be potential targets for controlling airway infection and inflammation in patients. If such effects can be synergized with chaperone-mediated regulation of CFTR biogenesis and quality control, more efficacious therapeutics will be developed to combat CF lung disease.
Keywords: CFTR, cystic fibrosis, drug discovery, F508del, immunity, inflammation, molecular chaperone, protein folding.
Graphical Abstract
Call for Papers in Thematic Issues
New drug therapy for eye diseases
Eyesight is one of the most critical senses, accounting for over 80% of our perceptions. Our quality of life might be significantly affected by eye disease, including glaucoma, diabetic retinopathy, dry eye, etc. Although the development of microinvasive ocular surgery reduces surgical complications and improves overall outcomes, medication therapy is ...read more
Related Journals

Anti-Cancer Agents in Medicinal Chemistry

Anti-Inflammatory & Anti-Allergy Agents in Medicinal Chemistry

Current Bioactive Compounds

Current Cancer Drug Targets

Combinatorial Chemistry & High Throughput Screening

Current Cancer Therapy Reviews

Current Diabetes Reviews

Current Drug Safety

Current Drug Therapy

Current Drug Delivery
Related Books

Drug Addiction Mechanisms in the Brain

Software and Programming Tools in Pharmaceutical Research

Objective Pharmaceutics: A Comprehensive Compilation of Questions and Answers for Pharmaceutics Exam Prep

Medicinal Chemistry of Drugs Affecting Cardiovascular and Endocrine Systems

Medicinal Plants, Phytomedicines and Traditional Herbal Remedies for Drug Discovery and Development against COVID-19

Advanced Pharmacy

Plant-derived Hepatoprotective Drugs
The Role of Chromenes in Drug Discovery and Development

New Avenues in Drug Discovery and Bioactive Natural Products

Practice and Re-Emergence of Herbal Medicine
 49
49 1
1
