





Abstract
Background: The recent epidemic of Zika virus infections in South and Latin America have raised serious concern on its ramifications for the population in the Americas and spread of the virus worldwide. The Zika virus disease is a relatively new phenomenon for which sufficient and comprehensive data and investigative reports have not been available to date.
Objective: To carry out a bioinformatics study of the available Zika virus genomic sequences to characterize the virus.
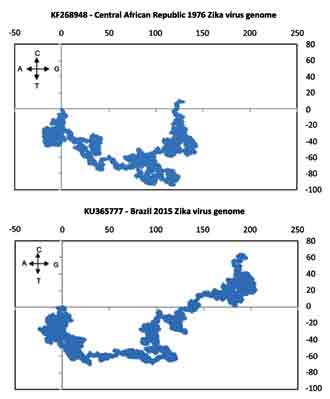
Method: 2D graphical representation method is used for visual rendering and compute sequence parameters and descriptors of the African and Asian-American groups of the Zika viruses to characterize the sequences. We also used MEGA5.2 and other software to compute various biological properties of interest like phylogenetic relationships, transition-transversion ratios, amino acid usage, codon usage bias and hydropathy index of the Zika genomes and virions.
Results: The phylogenetic relationships show that the African and Asian-American Zika virus genomes are grouped in two clades. The 2D plots of typical genomes of these types also show dramatic differences indicating that the gene sequences at the 5’-end coding regions for the structural proteins are rather strongly conserved. Among other characteristics, the transition/transversion ratio matrices for the sequences in each of the two clades show that analogous to the dengue virus, the transition rates are about 10 to 15 times the transversion rates.
Conclusion: These findings are important for computer-assisted approaches towards surveillance of emerging Zika virus strains as well as in the design of drugs and vaccines to combat the growth and spread of the Zika virus.
Keywords: Zika virus, Zika virus phylogeny, Zika virus characterization, African and Asian-American clades, 2D graphical representation, amino acid changes, cladewise transition-transversion ratios, Zika sequence descriptors.
Graphical Abstract
Related Journals

Anti-Cancer Agents in Medicinal Chemistry

Current Analytical Chemistry

Current Bioactive Compounds

Combinatorial Chemistry & High Throughput Screening

Current Medicinal Chemistry

Central Nervous System Agents in Medicinal Chemistry

Letters in Drug Design & Discovery

Current Drug Discovery Technologies

The Natural Products Journal

Current Catalysis
Related Books

Botanicals and Natural Bioactives: Prevention and Treatment of Diseases

Biosurfactants: A Boon to Healthcare, Agriculture & Environmental Sustainability

Frontiers in Computational Chemistry

Advances in Dye Degradation

Biological and Medical Significance of Chemical Elements

Frontiers In Medicinal Chemistry

Advances in Organic Synthesis

Frontiers in Natural Product Chemistry

Recent Advances in Analytical Techniques

Advanced Catalysts Based on Metal-organic Frameworks (Part 2)
 106
106 14
14 1
1 2
2